Recurrent Respiratory Infections with Malnutrition and Growth Delay — Beware of the Rare Pediatric Disease: Cystic Fibrosis (CF)

Some children seem to suffer from chronic respiratory problems that never fully resolve: recurrent pneumonia, persistent productive cough, poor weight gain, and difficulty gaining weight despite having a good appetite.
Many patients are repeatedly diagnosed with bronchitis, asthma, or simple malnutrition, yet their symptoms continue to recur. In some cases, the underlying cause may actually be Cystic Fibrosis (CF), a rare inherited disorder that is still underrecognized in many regions.
As a global pharmaceutical distributor focused on innovative therapies, rare diseases, and healthcare accessibility, DengYueMed introduces this often-overlooked pediatric disease to help more families understand its typical symptoms, diagnostic methods, and recent advances in treatment.
A Case of Cystic Fibrosis
Tom, an 8-year-old boy, had shown an unusual sign since early childhood: after sweating in summer, a fine layer of salt crystals would appear on his skin, and family members could even notice a distinctly salty taste when kissing him.
At the same time, he experienced recurrent coughing and purulent sputum production. He had been hospitalized multiple times for “pneumonia” and “bronchiectasis,” but anti-infective treatments provided only limited improvement.
More concerning findings included:
- A strong appetite but persistent failure to gain weight
- A BMI of only 13.5
- Frequent bulky, greasy, foul-smelling stools
- Significant growth and developmental delay compared with peers
Further evaluation revealed a sweat chloride concentration of 115 mmol/L, and genetic testing identified compound pathogenic mutations in the CFTR gene, leading to a definitive diagnosis of cystic fibrosis.
After receiving standardized treatment including pancreatic enzyme replacement, a high-calorie diet, airway clearance therapy, and nebulized therapy, Tom gradually gained weight and experienced a significant reduction in pulmonary infections.
This case highlights an important clinical message: when children present with recurrent respiratory infections together with malabsorption and growth failure, clinicians should maintain a high index of suspicion for cystic fibrosis.
What Is Cystic Fibrosis?
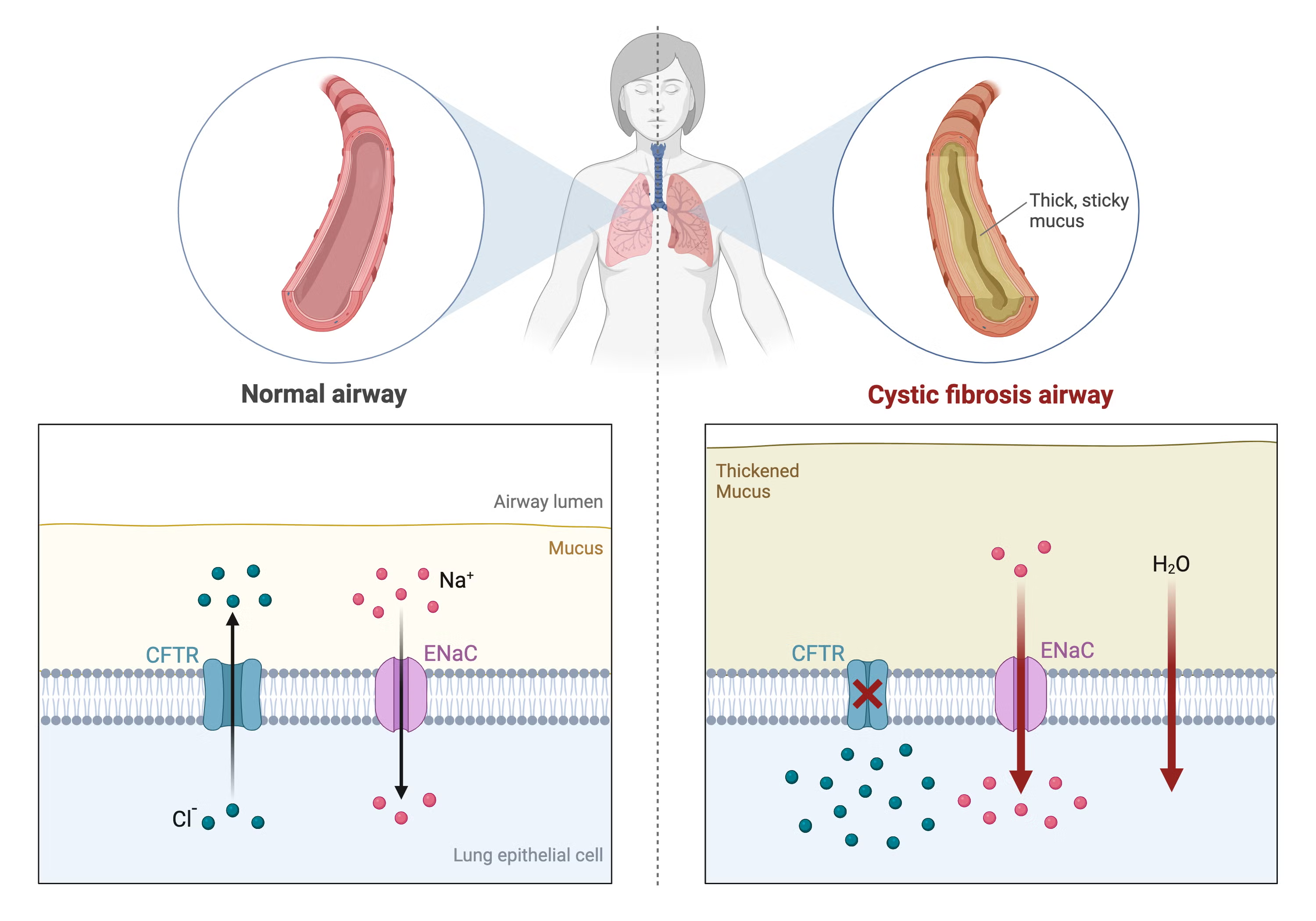
Cystic Fibrosis is an autosomal recessive genetic disorder caused by mutations in the CFTR gene.
The CFTR protein functions as a chloride ion channel located on epithelial cell membranes and plays a critical role in regulating salt and water transport.
When CFTR function is impaired, several pathological changes occur:
- Defective chloride and bicarbonate transport across epithelial membranes
- Reduced airway surface liquid
- Progressive dehydration and thickening of secretions
- Obstruction of ducts and chronic inflammation in multiple organs
These abnormalities affect the lungs, pancreas, gastrointestinal tract, liver, and reproductive system.
In the respiratory tract, thick and dehydrated mucus cannot be effectively cleared, allowing bacteria to persistently colonize the airways. This leads to recurrent infections and chronic inflammation. Over time, patients may develop bronchiectasis and progressive lung damage, which are major causes of morbidity in CF.
The “Salty Baby” Phenomenon: An Important Clinical Clue
Many children with CF have characteristically salty skin.
Because CFTR dysfunction impairs sodium chloride reabsorption in sweat glands, sweat contains abnormally high salt concentrations. Parents may therefore notice visible salt crystals on the skin after sweating or a salty taste when kissing their child.
Common manifestations include:
- Fine white salt crystals on the skin after sweating
- Noticeably salty skin
- Increased susceptibility to dehydration during summer
- Fatigue in hot weather
Although seemingly subtle, this feature is a classic clue to CF and forms the basis of the sweat chloride test used for diagnosis.
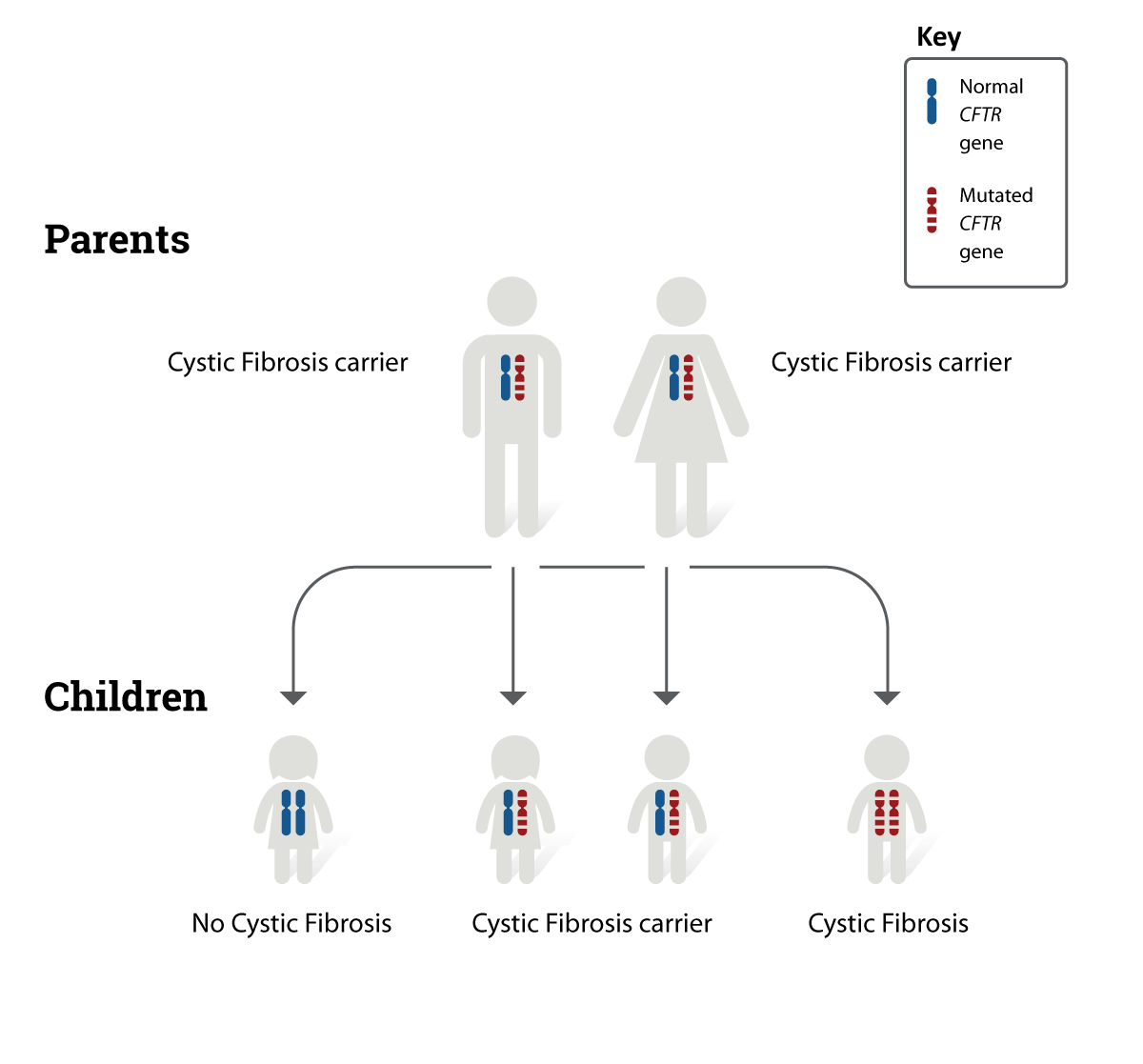
How Is Cystic Fibrosis Inherited?
CF is inherited in an autosomal recessive pattern. Most parents of affected children are asymptomatic carriers who each carry one pathogenic mutation.
When both parents are carriers:
- There is a 25% chance the child will have CF
- A 50% chance the child will be a carrier
- A 25% chance the child will be unaffected
More than 2,000 CFTR variants have been identified worldwide. Different mutations may influence disease severity and response to targeted therapies.
Typical Clinical Manifestations of Pediatric CF
1. Respiratory Manifestations
The respiratory system is the organ system most commonly affected in CF.
Symptoms often begin in infancy or early childhood and may include chronic cough, recurrent pneumonia, wheezing, and large amounts of purulent sputum.
As the disease progresses, patients may gradually develop:
- Bronchiectasis
- Digital clubbing
- Chest wall deformities
- Declining pulmonary function
Common respiratory pathogens include Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa.
Chronic colonization with Pseudomonas aeruginosa often indicates more advanced pulmonary disease.
2. Gastrointestinal and Nutritional Problems
Pancreatic insufficiency is common in CF, resulting in inadequate delivery of digestive enzymes into the intestine and impaired fat and protein absorption.
Affected children may present with:
- Steatorrhea
- Foul-smelling stools
- Malnutrition
- Growth retardation
- Failure to gain weight despite good appetite
Some newborns may initially present with meconium ileus.
3. Multisystem Involvement
CF can also affect multiple organ systems, including:
- Sinuses: chronic sinusitis and nasal polyps
- Liver: cholestasis and cirrhosis
- Endocrine system: CF-related diabetes (CFRD)
- Reproductive system: male infertility is common
Therefore, CF is considered a systemic disease rather than solely a pulmonary disorder.
How Is Cystic Fibrosis Diagnosed?
The diagnosis of CF requires integration of clinical findings, sweat testing, and genetic analysis.
Sweat Chloride Test (Gold Standard)
The sweat chloride test remains the classic diagnostic standard.
Interpretation is generally as follows:
- <30 mmol/L: usually normal
- 30–59 mmol/L: intermediate or inconclusive
- ≥60 mmol/L: highly suggestive of CF
Repeat testing is often recommended to reduce diagnostic error.
Genetic Testing
Identification of either homozygous CFTR pathogenic variants or compound heterozygous pathogenic variants further supports the diagnosis.
Genetic testing is also important for selecting appropriate precision-targeted therapies.
Conditions That May Be Confused with CF
Because CF is relatively uncommon in Asia, many patients are initially misdiagnosed.
| Disease | Key Differentiating Features |
|---|---|
| Primary ciliary dyskinesia (PCD) | Chronic cough and bronchiectasis may occur, but sweat chloride is normal |
| Primary immunodeficiency disorders | Recurrent infections often involve skin or gastrointestinal tract |
| Chronic diarrheal diseases | Predominantly gastrointestinal manifestations |
| Congenital pancreatic enzyme deficiency | Steatorrhea may occur, but chronic lung disease is usually absent |
Therefore, clinicians should maintain awareness of CF in children with recurrent respiratory infections and malnutrition.
How Is Cystic Fibrosis Treated?
Although CF is still considered incurable, advances in multidisciplinary management have significantly improved both survival and quality of life.
Core treatment goals include controlling infection, improving nutrition, preserving lung function, and slowing disease progression.
1. Airway Clearance Therapy
Airway clearance is a cornerstone of CF management and often requires lifelong daily adherence.
Common methods include:
- Active cycle of breathing techniques (ACBT)
- Chest percussion and postural drainage
- High-frequency chest wall oscillation
- Respiratory physiotherapy exercises
2. Nutritional Support and Pancreatic Enzyme Replacement
Children with CF typically require:
- High-calorie diets
- High-protein diets
- Supplementation of fat-soluble vitamins (A, D, E, and K)
Most patients also require pancreatic enzyme replacement therapy (PERT) to improve digestion and nutrient absorption.
3. Anti-Infective and Nebulized Therapy
Antibiotic therapy is selected based on sputum culture results. Some patients require long-term inhaled antibiotics or low-dose macrolide therapy to reduce pulmonary exacerbations.
Commonly used medications include:
- Tobramycin
- Azithromycin
4. CFTR Modulator Therapy: A Major Breakthrough in Precision Medicine
One of the most important recent advances in CF treatment is the development of CFTR modulators. Rather than only relieving symptoms, these therapies directly improve defective CFTR protein function.
Representative agents include:
- Ivacaftor
- Lumacaftor
- Tezacaftor
- Elexacaftor
Many patients experience substantial improvements in pulmonary function, nutritional status, and quality of life following treatment.
However, because different mutations respond to different therapies, and due to the high cost of treatment, global accessibility remains unequal.
Conclusion
If a child experiences recurrent respiratory infections, persistent pneumonia or bronchiectasis, together with malnutrition, poor growth, greasy foul-smelling stools, or unusually salty skin, the possibility of Cystic Fibrosis should not be overlooked. Early diagnosis and standardized treatment are critical for improving long-term outcomes.
With the development of CFTR-targeted therapies, CF management is entering the era of precision medicine, offering improved disease control and quality of life for increasing numbers of patients.
Global pharmaceutical distributor DengYueMed continues to focus on rare diseases and innovative therapies worldwide, providing updates on international drug supply and emerging treatment advances to help patients and healthcare stakeholders better understand global therapeutic developments and pharmaceutical information.
Related Posts
- When ADCs Achieve a 50% Objective Response Rate: How Technological Innovation Translates into Patient Benefit
- Mapping China's RAS Pipeline Landscape: From “Undruggable” Target to a New Frontier in Oncology Innovation
- Why Are ADC Drugs Remaining So Hot? A Comprehensive Analysis of the Global Cancer Innovation Landscape