New Advances in Gaucher Disease Classification: Clinical Characteristics of Type I, Type II, Type III, and Rare Subtypes
Introduction
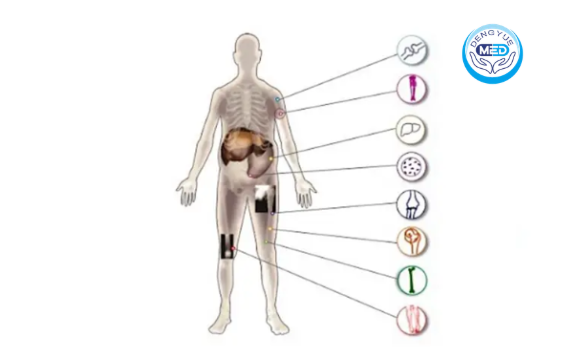
Gaucher Disease (GD) is a rare autosomal recessive lysosomal storage disorder caused by mutations in the GBA gene, resulting in reduced or deficient glucocerebrosidase activity. Consequently, glucocerebroside accumulates abnormally within macrophages, forming characteristic “Gaucher cells.”
These abnormal cells can infiltrate the liver, spleen, bones, lungs, bone marrow, and nervous system, leading to multisystem damage.
As precision medicine continues to evolve, the classification system of Gaucher disease is becoming increasingly refined. Modern clinical research now recognizes not only the traditional Type I, Type II, and Type III forms, but also several rare subtypes with unique genetic and clinical characteristics.
Basis of Gaucher Disease Classification
Traditionally, Gaucher disease has been classified according to the presence and progression of neurological involvement:
- Type I: Non-neuronopathic type
- Type II: Acute neuronopathic type
- Type III: Chronic or subacute neuronopathic type
In recent years, clinicians have further identified rare forms including the perinatal lethal subtype and cardiovascular Gaucher disease, helping establish a more precise disease stratification framework.
Research-focused medical platforms such as DengYueMed are increasingly covering developments in genetic profiling, disease progression, and emerging therapies for Gaucher disease.
I. Type I Gaucher Disease: The Most Common Non-Neuronopathic Form
1. Clinical Features
Type I Gaucher disease accounts for over 90% of all cases and is considered the most common subtype.
The hallmark characteristic is the absence of primary central nervous system involvement.
Symptoms may appear during childhood, adolescence, or adulthood, and disease progression is usually slow.
Major Clinical Manifestations
- Hepatosplenomegaly (especially marked splenomegaly)
- Anemia and thrombocytopenia
- Bone pain and osteoporosis
- Pathological fractures
- Growth retardation
- Easy bruising and chronic fatigue
Some patients may additionally develop:
- Pulmonary involvement
- Gallstones
- Immune dysregulation
Severe skeletal complications remain one of the major factors affecting long-term quality of life.
2. New Insights in Recent Years
Historically, Type I Gaucher disease was considered completely non-neuronopathic.
However, recent studies suggest that certain patients may later develop:
- Peripheral neuropathy
- Parkinsonian manifestations
- Cognitive decline
- Increased neurodegenerative risk
Notably, mutations in the GBA gene are now recognized as important genetic risk factors for Parkinson’s disease.

3. Treatment Progress
Most Type I patients respond well to:
- Enzyme replacement therapy (ERT)
- Substrate reduction therapy (SRT)
- Long-term individualized disease management
With appropriate treatment, many patients experience substantial improvement in:
- Liver and spleen size
- Hematologic parameters
- Bone pain
- Daily functioning
II. Type II Gaucher Disease: Rapidly Progressive Acute Neuronopathic Form
1. Clinical Features
Type II Gaucher disease is among the rarest and most severe forms.
Symptoms typically appear during infancy and rapidly progress.
Common Manifestations
- Feeding difficulties
- Abnormal muscle tone
- Opisthotonos
- Seizures
- Dysphagia
- Respiratory distress
- Abnormal eye movements
Most affected children die before 2–3 years of age.
2. Neurological Characteristics
Neurological deterioration is the defining feature of Type II disease.
Major Neurological Findings
- Brainstem dysfunction
- Severe motor impairment
- Progressive neurodegeneration
- Extensive brain injury
Because conventional enzyme replacement therapy cannot effectively cross the blood-brain barrier, neurological symptoms remain difficult to treat.
3. Current Research Hotspots
Emerging therapeutic research areas include:
- Gene therapy
- Small-molecule chaperone therapy
- Blood-brain barrier delivery systems
- Neuro-targeted therapies
Although no curative treatment currently exists, ongoing advances in gene editing and brain-directed therapeutics may significantly improve outcomes in the future.
III. Type III Gaucher Disease: Intermediate Between Type I and Type II
1. Clinical Features
Type III Gaucher disease is classified as a chronic neuronopathic form with intermediate severity.
Patients often develop symptoms during childhood but survive significantly longer than those with Type II disease.
Systemic Manifestations
- Hepatosplenomegaly
- Skeletal disease
- Hematologic abnormalities
Neurological Manifestations
- Oculomotor abnormalities
- Ataxia
- Seizures
- Cognitive decline
- Progressive neurological dysfunction
2. Subclassification of Type III Disease
Type IIIa
Primarily characterized by progressive neurodegeneration:
- Ataxia
- Dementia
- Progressive neurological decline
Type IIIb
Dominated by visceral and skeletal involvement:
- Massive hepatosplenomegaly
- Severe bone disease
- Relatively mild neurological impairment
Type IIIc
A unique subtype associated with:
- Aortic calcification
- Cardiac valvular fibrosis
- Supranuclear gaze palsy
Because of severe cardiovascular complications, Type IIIc has gained increasing clinical attention.
IV. Rare Subtypes: Perinatal Lethal Form and Cardiovascular Type
1. Perinatal Lethal Form
This represents one of the most severe forms of Gaucher disease.
Typical Features
- Hydrops fetalis
- Severe skin abnormalities
- Respiratory failure
- Severe neurological impairment
Most infants die shortly after birth.
2. Cardiovascular Gaucher Disease
Major manifestations include:
- Aortic valve calcification
- Cardiac valvular fibrosis
- Cardiac dysfunction
Some patients may also exhibit mild neurological and oculomotor abnormalities.
Recent studies suggest strong associations between specific GBA mutations and cardiovascular phenotypes.
V. Comparison of Clinical Characteristics Among Different Types
| Type | Neurological Involvement | Age of Onset | Major Manifestations | Prognosis |
|---|---|---|---|---|
| Type I | No significant primary CNS involvement | Childhood to adulthood | Hepatosplenomegaly, anemia, skeletal disease | Relatively good |
| Type II | Severe and rapidly progressive | Infancy | Seizures, brainstem dysfunction, dysphagia | Very poor |
| Type III | Chronic progressive involvement | Childhood | Neurological symptoms with skeletal and visceral disease | Intermediate |
| Perinatal lethal form | Extremely severe | Fetal period | Hydrops fetalis, respiratory failure | Extremely poor |
| Cardiovascular type | Mild possible involvement | Childhood | Valvular calcification, aortic disease | Depends on cardiac damage |
Conclusion
Although Gaucher disease is classified as a rare disease, its clinical heterogeneity is remarkably complex.
From the chronic visceral manifestations of Type I disease to the rapid neurodegeneration of Type II disease, the mixed neurological and skeletal involvement of Type III disease, and the increasingly recognized rare subtypes, medical understanding of Gaucher disease continues to evolve.
The refinement of classification systems has improved diagnostic precision while also opening new directions for:
- Individualized therapy
- Precision medicine
- Gene-targeted interventions
- Brain-directed treatments
With continued advances in gene therapy, neurological drug delivery, and molecular medicine, the long-term prognosis and quality of life of patients with Gaucher disease are expected to improve substantially in the future.
Related Posts
- When ADCs Achieve a 50% Objective Response Rate: How Technological Innovation Translates into Patient Benefit
- Mapping China's RAS Pipeline Landscape: From “Undruggable” Target to a New Frontier in Oncology Innovation
- Why Are ADC Drugs Remaining So Hot? A Comprehensive Analysis of the Global Cancer Innovation Landscape